




精选内容
-
矫大夫您好,我是视神经脊髓炎,赛可平以前是分散片,后来换成了胶囊,血药浓度和以前不一样有影响吗?

 矫毓娟医生的科普号
矫毓娟医生的科普号 2024年10月29日
2024年10月29日 58
58
 0
0
 1
1
-
神经免疫问题不容忽视,规范合理很重要
周红雨:视神经脊髓炎谱系疾病诊疗进展详解|2023年中国罕见病大会原创 医学界报道组 医学界罕见病频道 2023-10-3119:31 重庆 11人听过仅供医学专业人士阅读参考视神经脊髓炎谱系疾病(NMOSD)鉴别诊断、治疗最新进展的全面介绍~视神经脊髓炎谱系疾病(NMOSD)是一种自身免疫介导的以视神经和脊髓受累为主的中枢神经系统炎性脱髓鞘疾病,反复存在的AQP4-IgG抗体是其典型特征。该疾病具有高复发、高致残的特点,可导致视力丧失和瘫痪[1],预后极差。NMOSD病情”来势汹汹“,及时诊疗尤为重要。现阶段医学界在NMOSD诊疗领域有哪些新进展、新观点呢?不久前,中国罕见病联盟、中国红十字基金会、全国罕见病诊疗协作网办公室等共同主办的“2023年中国罕见病大会”在京召开,四川大学华西医院神经内科主任医师周红雨教授对NMOSD的诊疗进展进行了全面解读,并接受了“医学界”专访,让我们共同来学习~,时长09:NMOSD概述临床症状NMOSD首次发病见于各年龄阶段,好发于青壮年,女性居多。临床上多以严重的视神经炎(ON)和纵向延伸的长节段横贯性脊炎(LETM)为特征表现,6组NMOSD的核心临床症候分别为视神经炎、急性脊髓炎、极后区综合征、急性脑干综合征、急性间脑综合征和大脑综合征。NMOSD为高复发、高致残性疾病(图1);常与一些自身免疫疾病如干燥综合征、系统性红斑狼疮、桥本氏病等发生共病现象。图1:NMOSD的高复发、高致残性致病机制目前,整体上认为AQP4-IgG是NMOSD致病性抗体。血清AQP4-IgG主要在外周由浆细胞和浆母细胞产生,并通过内皮胞吞作用或相对血脑屏障通透性或损伤区城穿透进CNS,导致AQP4-IgG与星形胶质细胞上的AQP4蛋白相结合。抗体依赖性星形胶质细胞损伤涉及CDC、CDCC和ADCC等机制,并进一步导致炎症反应、少突胶质细胞丢失、脱髓鞘和神经元丢失(图2)。图2:NMOSD发病机制遗传影响因素(1)HLA(2)免疫调节基因NMOSD发病风险增加相关基因:CD40、STAT4、GTF2IRD1-GTF21、TNFSF4、CD58;NMOSD发病风险减少相关基因:IRAK1。NMOSD的诊断谈及对于NMOSD患者的诊断,周红雨教授指出,尽管NMOSD与多发性硬化(MS)在临床表现、影像学具有诸多相似之处,但两者的诊断却“大相径庭”。MS的诊断基于影像学寡克隆区带,其病灶具有空间多发性,且脊髓MRI多呈短节段病灶。而NMOSD的诊断(如图3)以“病史+核心临床症候+影像特征+生物标记物”为基本依据,以AQP4-IgG作为分层。AQP4-IgG对于NMOSD的诊断具有较高的特异性,达90%-95%。因而NMOSD的诊断基于AQP4-IgG检测,若检测为阳性,结合6组核心症状(视神经炎、急性脊髓炎、极后区综合征、急性脑干综合征、急性间脑综合征和大脑综合征)中的任意一组,便可确诊为NMOSD;但也有少部分患者AQP4-IgG呈阴性,对于该类患者需参考其他亚临床及免疫学证据做出诊断。除此之外,我们还需排除其他疾病如MS,以提高诊断的准确性。图3:NMOSD诊断标准关于NMOSD患者的AQP4-IgG检测,一项小型研究表明,AQP4-IgG可能在NMOSD发作前16年就存在。因此,为了确定NMOSD患者的症状前血清中是否存在AQP4-IgG,一项基于美国国防部血清储备库(DoDSR)样本的前瞻性研究应运而生。该研究选取了64例确诊的NMOSD患者,并按比(1:1:1)选取了年龄性别相匹配的健康和MS对照者(图4)。结果发现(图5),健康和MS对照组中,参与者的AQP4-IgG检测都呈阴性,而50%的NMOSD患者在发病前1200天就已经出现了AQP4-IgG阳性。如此看来,部分患者即使AQP4-IgG阳性,但其视神经系统却无损害,因而也无法确诊为NMOSD。甚至有些AQP4-IgG阳性的患者终生不发病。只有当体内补体激活,细胞毒性抗体等促进急剧的炎症反应,损害视神经系统4:5:研究结果曲线NMOSD的治疗AQP4抗体与预后传统免疫抑制剂治疗中位3.7年后,32%的患者AQP4抗体可以转阴。抗体转阴后,患者年复发率明显降低,且转阴时间越早,复发风险越低。发病年龄越小(50岁以下)、首次发作累积多个部位、治疗期间AQP4抗体滴度不变或升高的患者未来复发风险较高。AQP4抗体滴度动态变化可作为预测复发和反映疗效的指标。治疗关于NMOSD的治疗,周红雨教授指出,急性期治疗和序贯治疗需“各司其职”,缺一不可。急性期治疗急性发作通常会引起中度至重度的炎症性损伤,每次发作都会导致残疾的累积,且平均神经功能状况评估量表评分(EDSS)增加约1分。NMOSD任何一次临床发作均有可能带来不可逆性损伤,其残障主要归因于发作后视觉功能缺损的累积,因此,及时治疗在急性发作期显得尤为重要。▌ 糖皮质激素激素脉冲疗法(IVMP)仅对83.8%的NMOSD患者有效,包括完全有效(17%)和部分有效(65.4%)。但多次接受IVMP的NMOSD患者,其疗效可能显著降低。▌ 血浆置换(PLEX)该疗法旨在是去除血液中的AQP4抗体,细胞因子和补体,以避免进一步的炎症反应从而发病。2013年的一项研究显示,50%的患者在PLEX一个疗程结束后,病情得到改善,78%的患者在6个月内得到了改善,且平均AQP4抗体滴度降低了85%。经IVMP结合PLEX治疗的NMOSD-ON患者的平均最终视力为20/50,而单独接受IVMP或PLEX的患者的平均最终视力为20/400,换句话说,PLEX与IVMP的结合治疗比IVMP单一疗法对重度脊髓炎和NMOSD-ON患者的疗效更佳。研究表明(图6),NMOSD急性期发作及时干预有助于病情完全恢复。因此,NMOSD患者处于急性期,应立即就诊,且首选激素脉冲疗法结合血浆置换。图6结果表明急性期的PLEX有助于病情序贯治疗对于AQP4-IgG阳性以及AQP4-IgG阴性复发病程的患者,一经诊断应尽早开始序贯治疗,并坚持长程治疗,以预防复发。▌ 生物制剂:萨特利珠单抗、利妥昔单抗、伊纳利珠单抗、依库珠单抗、托珠单抗传统治疗大多使用利妥昔单抗。此外,欧洲一项多中心的全球临床3期试验——CHAMPION-NMOSD试验,通过评估AQP-4抗体阳性的NMOSD成人患者使用依库珠单抗(长效C5体抑制剂)的疗效,已证实该药物的有效性与安全性,相信不久该药物将上市,未来我们的药物选择也将更多。▌ 免疫抑制剂:吗替麦考酚酯、硫唑嘌呤、甲氨蝶呤、他克莫司、环磷酰胺、米托蒽醌一项关于视力残疾的预后研究(图7),通过对644例NMOSD患者随访10年,发现其中161例患者出现单眼视力残疾,且NMOSD视力残疾危险因素包括首发年龄(非线性),首发视神经炎(HR=2.91),AQP4抗体阳性(HR=2.98),首次严重发作(HR=1.93),免疫治疗前ARR高(HR=1.85),由此得出免疫抑制剂治疗可显著降低视力残疾风险(HR=0.1)。而另一项关于运动残疾的预后研究表明,免疫抑制剂(MMF)、硫唑嘌呤(AZA)治疗可显著延缓NMOSD患者发生运动残疾时间,而单纯激素治疗不能改善NMOSD患者残疾结局。因而免疫抑制剂治疗也可显著降低NMOSD运动残图7视图8 运动残疾的预后研究综上所述,免疫抑制剂治疗可以很好地改善NMOSD患者的预后——降低视力残疾和运动残疾的风险。小结与展望总的来说,NMOSD是一个高复发、高致残的疾病,且致病“罪魁祸首”AQP4抗体的出现很可能早于我们目前的认识。对于NMOSD的准确诊断,AQP4抗体滴度的监测和随访至关重要。明确诊断后进行及时且高效的预防治疗是降低残疾风险的关键,可有效改善预后,提高患者的生活质量。关于预防NMOSD的复发治疗,周红雨教授强调,以往临床使用激素与传统的免疫抑制剂结合,而现阶段随着对单抗类药物的研究,其比传统疗法更加的有效性与安全性。近两年,国内单抗类药物逐步上市,且基本上都申入医保,相信能为NMOSD患者带来更佳的治疗效果!周红雨教授也期望有更多的年轻医生和基层医生参与到NMOSD等罕见病的治疗与长期管理当中,为罕见病患者增添生命色彩!参考文献:[1]中国免疫学会神经免疫分会.中国神经免疫学和神经病学杂志,2021,28(6):423-436.周红雨 教授四川大学华西医院关注“医学界罕见病频道”,这里有大咖经验分享、罕见病诊疗知识、前沿新药及政策资讯,点击文末阅读原文或长按识本文来源:医学界罕见病"医学界"力求所发表内容专业、可靠,但不对内容的准确性做出承诺;请相关各方在采用或以此作为决策依据时另行核查。投稿邮箱:hanjianbing@yxj.org.c医学界罕见病频道
复旦大学附属浦东医院科普号 2024年03月20日82
0
64
2024年03月20日82
0
64
-
世界视神经脊髓炎日:作为患者家属,我们可以做什么?
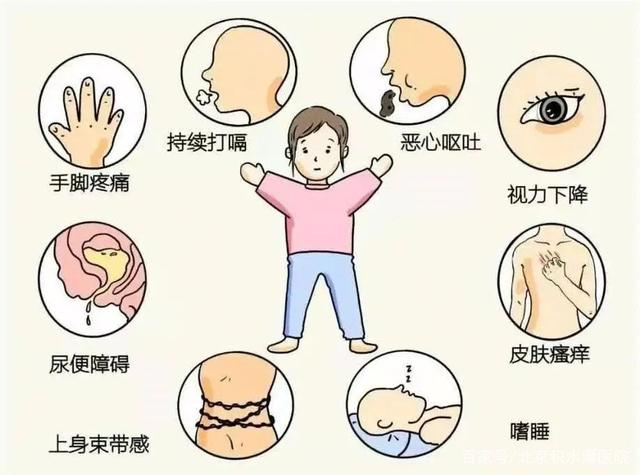
2023年10月26日,是“视神经脊髓炎”(NMO,2007年后也称为视神经脊髓炎谱系病NMOSD)命名129周年,但是,了解此疾病的人们并不多,能正确面对此疾病的患者及家属也很少。但此疾病亚洲人群高发,年轻女性更易发,有易复发、高致残性特点,可能会给家庭、社会带来较大负担。因此,需要有更多的人去了解它从而正确应对哦。NMOSD的前身,名为视神经脊髓炎(NMO),之前一直被认为是多发性硬化(MS)的一种亚型,随着AQP4抗体的被发现,在2004年被明确是一种独立疾病。到了2015年,国际上明确了成人NMOSD的诊断标准。终于,100多年后,此疾病可以被明确诊断了。它是一种罕见的神经免疫性疾病,因为免疫系统故障,会影响身体多个部位而出现视物模糊、肢体麻木乏力、大小便功能障碍、恶心呕吐等多种症状。它可能会像感冒一样,抵抗力减退时,会发作一次;但它又不像感冒那样简单,需要长疗程治疗。若应对不好,可能会导致残疾甚至死亡。因此,除了患者自己,NMOSD患者家属,也是一起战胜克服此疾病的主力军。1、和患者一起了解此疾病、共同面对此疾病。NMOSD是一种可能会复发,需要长期规范治疗的疾病,但它不是癌症,是可治的,积极面对、规范治疗,就可以克服疾病回归正常,不影响正常工作生活。2、与患者做好沟通。在疾病发作时,尽量不要过度询问症状恢复情况,安心陪护、适当关心,不引导焦虑情绪,要给予积极心态。要知道,患者本身已经很紧张焦虑了,但此疾病急不来,一起慢慢面对才是硬道理。3、与患者一起积极正确应对病痛。患者病情急性发作后,立即就医治疗,尽量早治疗早恢复。急性期治疗后,肢体功能障碍可能不会立即完全恢复,给予患者积极的心态早期康复锻炼,同时遵医嘱随访调整药物。4、特殊人群的特殊关怀。1)育龄期女性NMOSD易发病于年轻女性,但它并不是遗传性疾病,疾病本身对胎儿也没有影响,因此NMOSD患者可以怀孕。但是,作为家属,可不能太过着急哦。保小的同时,还需要保大。因此,育龄期NMOSD患者,若有备孕计划,需要与专业医生沟通,病情良好控制后再选择备孕。妊娠和产后可能有复发风险,但不要过于紧张,怀孕及产后注意休息,避免疲劳、情绪紧张及感染,在专业医生指导下,在不同阶段调整用药后,NMOSD患者同样能顺利怀孕生娃。2)老年患者NMOSD主要是因为免疫异常引起,而老年患者本身免疫力低下,可能有较多合并疾病,因此在长期用药及预防方面,需要有更多注意。比如,治疗与日常生活期间更需要注意预防感染、注意保暖;老年患者本身易骨质疏松,部分情况下需要使用激素,就可能更容易发生骨折等,因此,老年患者需要更注意补钙、避免摔跤等。另外,老年患者的功能残障恢复相对会慢些,家属更需要有耐心,选择规范的康复方式鼓励功能恢复,同时注意加强护理,预防比如感染、褥疮等并发症的发生。总之,认识疾病、正确面对、有效沟通、积极心态,我们一起努力,终能一起战胜病痛。最后,上海交通大学医学院附属仁济医院神经内科一直是为NMOSD患者开辟诊疗绿色通道,长期规律随访,积极探索安全有效的治疗方式。一起积极面对,终能点亮你的生命色彩!
 丁婕医生的科普号
丁婕医生的科普号 2023年10月26日443
0
0
2023年10月26日443
0
0
-
2023年视神经脊髓炎谱系疾病最新的治疗推荐
2023年9月欧洲视神经脊髓炎谱系疾病(NMOSD)工作组在神经病学杂志上发表了NMOSD的最新治疗推荐。一、NMOSD急性发作时的治疗:急性发作时治疗越早越好;治疗方案①:甲泼尼龙1g/天静脉输注,3-5天;如果无明显恢复,及早进行血浆置换或免疫吸附治疗5-10次(每天或隔日一次);如果有效,激素减量,口服维持3-6月;②既往发作甲泼尼龙效果不好患者、既往发作血浆置换或免疫吸附效果好的患者、或者严重脊髓炎患者可首选血浆置换或免疫吸附治疗5-10次;如果治疗有效,口服激素维持3-6月;如果治疗效果不好,甲泼尼龙1g/天静脉输注,3-5天,随后口服激素3-6月;③严重发作,联合甲泼尼龙和血浆置换/免疫吸附治疗;尽管静脉注射用免疫球蛋白可能有效,但并未推荐。二、预防复发的治疗①AQP4抗体阳性的NMOSD患者首次发作后应给与长期免疫抑制药物治疗预防复发;对患者既往使用免疫抑制药物病情稳定的患者,又无明显副作用,不需要改变治疗药物;传统的免疫抑制剂如硫唑嘌呤、吗替麦考酚酸、口服激素可以使用,但作为非首选药物(二线药物);不主张单独小剂量激素口服预防复发;单克隆抗体药物如依库利珠单抗、依库珠单抗、伊奈利珠单抗、利妥昔单抗、沙特利珠单抗治疗效果优于传统免疫抑制剂,均可作为AQP4抗体阳性NMOSD患者的一线预防药物。但需要根据患者个体情况如发作严重性、并存病、妊娠计划、家庭财政情况等选择;②AQP4抗体阴性的NMOSD患者首次严重发作或第二次发作后建议长期免疫抑制药物治疗预防复发;利妥昔单抗和经典传统免疫抑制剂(硫唑嘌呤吗替麦考酚酸)均可作为一线药物;三、治疗失败患者药物选择治疗失败定义:治疗过程中(有效药物剂量和有效作用时间内)仍出现严重发作或者发生药物严重副作用;①硫唑嘌呤、吗替麦考酚酸治疗失败患者应转换为单克隆抗体治疗;②单克隆抗体治疗失败可换为另一种作用机制不同的单抗药物;③转换药物过程中可给与小剂量激素口服等待新药物发挥作用;四、治疗疗程:AQP4抗体阳性NMOSD患者建议长期免疫治疗;AQP4抗体阴性NMOSD患者稳定至少5年;五、妊娠:妊娠期NMOSD患者应早期根据妊娠计划选择免疫治疗;不应因准备妊娠中断免疫治疗;硫唑嘌呤、利妥昔单抗、依库利珠单抗在妊娠期间可继续应用,但需要密切监测副作用;
任士卿医生的科普号 2023年09月15日540
0
2
2023年09月15日540
0
2
-
矫主任,您好,视神经脊髓炎谱系疾病,吃赛可平两片激素,最近几个月吃着匹伐他丁低密度胆固醇还是有点高
 矫毓娟医生的科普号2023年08月30日37
0
0
矫毓娟医生的科普号2023年08月30日37
0
0
-
视神经脊髓炎导致的视神经萎缩用鼠神经生长因子有必要吗
矫毓娟医生的科普号2023年08月16日38
0
0
-
因为坚持,和视神经脊髓炎谱系病痛说拜拜!
杨先生,46岁,双下肢活动不利18年、双眼视力下降、二便障碍7月,诊断为视神经脊髓炎谱系疾病,饱受疾病痛苦但因各种原因未规范诊治随访,首次于我院就诊时,已因疾病反复发作、进行性加重,导致长期卧床,留置导尿,严重压疮感染。中青年男性,已生活无法自理、无法正常进入社会工作。 刘女士,43岁,同样得了视神经脊髓炎谱系疾病,同样经历了数年数次发作,后遗视力稍有下降、肢体麻木。期间经历过因为免疫抑制剂不耐受而感染高热等副反应,但患者及家属以积极心态应对疾病,定期规律复诊随访,遵医嘱调整预防复发药物,目前已3年未复发,生活不受影响,每次门诊随访乐观开朗,已与随访医生成为好朋友。同样经受了视神经脊髓炎谱系疾病(NMOSD)这一高复发、高致残性的好发于中青年的神经系统疾患,两位患者却有截然相反的结局,其中只因刘女士坚持一件事,那就是正视疾病、定期规律随访、遵医嘱用药,因为坚持这件事,她不仅与医生成了好朋友,还与病痛说拜拜。什么是视神经脊髓炎谱系疾病?视神经脊髓炎谱系疾病(NMOSD)是一种中枢神经系统自身免疫性疾病,位列国家《第一批罕见病目录》。因为致病抗体水通道蛋白4(AQP4)特异性自身抗体分布的特殊性,可以累及视神经、脑、脊髓等多个部位损伤,主要有视力下降、肢体麻木、瘫痪、排尿困难、便秘、呃逆等症状。有高复发、高致残性,90%以上患者为多时相病程,60%患者在1年内复发,90%患者在3年内复发,多数患者遗留有一定程度的功能残疾,严重影响患友们的正常生活。不幸患病,不是我们自身能控制的,但正确对待、与疾病友好共处是我们可以自己来决定的。正视疾病,正确治疗随访,也可以有很好的预后。如何规范治疗,随访应该关注哪些指标?首先,一旦有上述可疑症状出现,应及时与医生取得联系、及时医院就诊。上海交通大学医学院附属仁济医院神经内科由管阳太主任为领衔的神经免疫团队,有着丰富的临床诊治经验和专业的技术水平。一旦有急性发作的神经免疫性疾病患者就诊,比如视神经脊髓炎谱系疾病急性复发,会“绿色通道”优先收住院,及时诊断及治疗。以尽量尽早减少神经系统损伤、减少失明瘫痪等严重后果,改善预后。NMOSD的治疗分为急性期和缓解期治疗,急性期包括大剂量激素冲击、丙种球蛋白、血浆置换治疗,缓解期主要有吗替麦考酚酯、硫唑嘌呤、利妥昔单抗等预防复发的药物。由于NMOSD疾病本身高复发性,预防复发用药起效时间不同、且患者有不同耐受性、部分患者可能有一定的副作用,因此,急性期治疗好转出院后,仍需定期规律复诊随访,主要关注的指标就是疗效和安全性。出院后,需要进行比如血常规、肝肾功能等副作用指标的定期监测、还有临床症状评分、淋巴细胞亚群、CD20、AQP4抗体等血免疫指标、磁共振影像等体现疗效的监测。随访期限根据不同患者特殊性决定,刚用药时需要短期两周左右频繁复诊复查,后期稳定后,仍需至少半年一次随访。根据疗效和安全性指标的监测,会调整用药剂量等,以达到最好的预后。我科专业医生会定期随访,更需要患者对随访重要性的高度认识及配合。希望更多的NMOSD患友,能够像刘女士一样,虽然得此严重罕见病,但并不可怕。与随访医生一起,以乐观积极心态正视疾病,定期规律随访,与疾病痛苦说拜拜!
 丁婕医生的科普号2023年07月28日402
0
2
丁婕医生的科普号2023年07月28日402
0
2
-
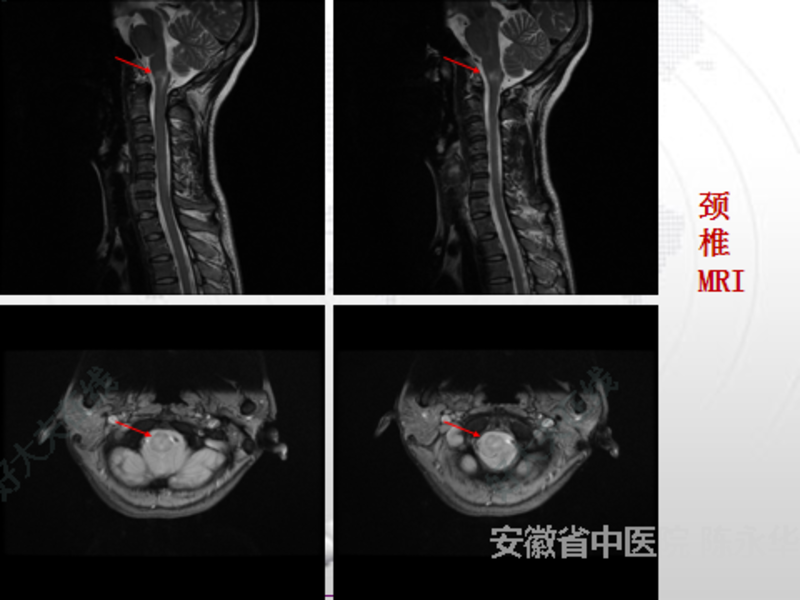
视神经脊髓炎谱系疾病
 陈永华医生的科普号2023年07月04日118
0
6
陈永华医生的科普号2023年07月04日118
0
6
-
视神经脊髓炎谱系病高复发风险因素
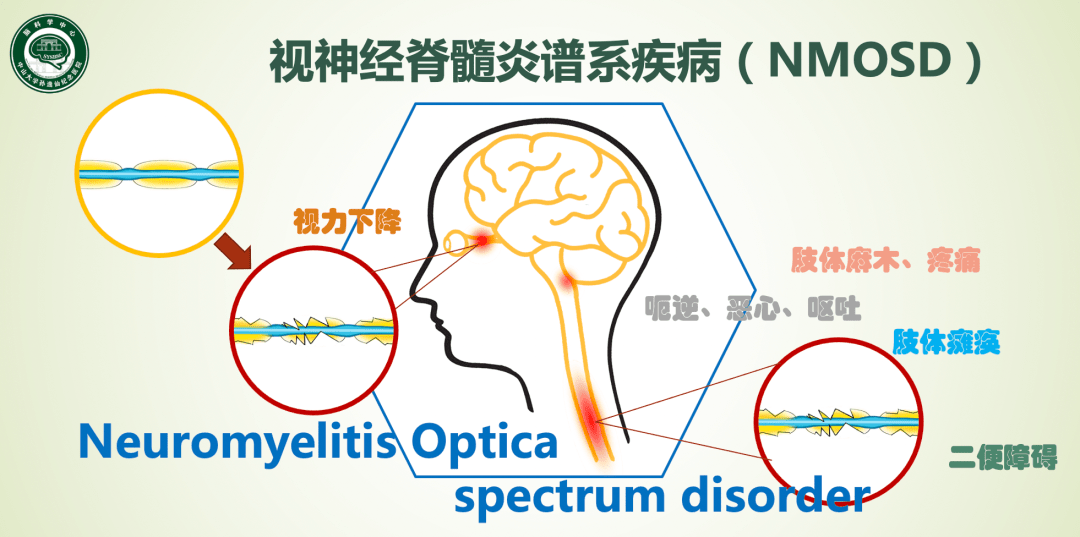
病史:女性,感染、妊娠晚期和产后、未治疗患者接种疫苗、存在伴发疾病临床特征:发作1年内、疼痛性强直性痉挛(PTS症状)、瘙痒实验室指标:高AQP4-IgG滴度、GFAP水平及IL-6水平,高胆固醇/甘油三酯血症影像学:长节段脊髓炎、头颅磁共振存在强化、髓质病变!治疗:未使用维持治疗、口服激素时间小于半年、既往相同治疗有发作!
 张艳林医生的科普号
张艳林医生的科普号 2023年06月17日80
0
0
2023年06月17日80
0
0
-
你好娇主任。孩子 11 岁。视神经脊髓炎。可以打伊奈丽珠单抗吗
矫毓娟医生的科普号2023年06月07日41
0
0
相关科普号
胡兵医生的科普号
胡兵 主任医师
上海中医药大学附属龙华医院
肿瘤科
6975粉丝19万阅读
秦伟医生的科普号
秦伟 主任医师
首都医科大学附属北京朝阳医院
神经内科
104粉丝9157阅读

张力医生的科普号
张力 主治医师
上海市第一人民医院(南部)
神经内科
322粉丝26.1万阅读
-

 推荐热度5.0李海峰 主任医师宣武医院 神经内科
推荐热度5.0李海峰 主任医师宣武医院 神经内科重症肌无力 76票
急性脊髓炎 15票
多发性硬化 11票
擅长:神经肌肉接头疾病(重症肌无力等),中枢神经脱髓鞘疾病(多发性硬化,视神经脊髓炎等),炎症性周围神经病,其他神经免疫疾病,头疼,脑血管病 -
 推荐热度4.7张伟赫 主任医师中日医院 神经科
推荐热度4.7张伟赫 主任医师中日医院 神经科急性脊髓炎 11票
重症肌无力 9票
脑梗塞 2票
擅长:视神经脊髓炎谱系疾病、脊髓炎/脊髓病、多发性硬化、重症肌无力、视神经炎、小脑共济失调(副肿瘤性小脑变性,谷蛋白共济失调等)、桥本脑病、副肿瘤性边缘叶脑炎、自身免疫性脑炎、脑炎、急慢性格林巴利综合症等神经疾病和神经免疫疾病的诊断和治疗 -

 推荐热度4.6赵桂宪 副主任医师复旦大学附属华山医院 神经内科
推荐热度4.6赵桂宪 副主任医师复旦大学附属华山医院 神经内科中枢神经系统脱髓鞘疾病 24票
脑炎 13票
急性脊髓炎 13票
擅长:擅长中枢神经系统脱髓鞘疾病,多发性硬化,视神经脊髓炎、脑白质病等,专长“颅内多发病变”的诊断和鉴别诊断,对周围神经病和神经系统遗传变性病也有一些自己的体会