




精选内容
-
儿童康复治疗选用什么方法?家长需要了解的!
康复治疗选用什么方法?家长需要了解的!从事小神经内科专业,主要诊治儿童脑发育方面疾病,开展儿童早期脑发育保健、预防脑发育障碍以及共患病、早期干预和康复治疗的近40年,总结以往经验,结合本人大量长期随访病例预后资料,以及国内外相关的临床研究结果,对常用的儿童早期干预和康复治疗的方法总结进行归纳,主要分为以下几类,以供从事这方面的专业人员和有需要了解这方面知识的家长参考。针对发育落后孩子的早期干预、康复治疗的主要方法:一、针对性的治疗方法,主要指的是一对一的教育训练1.主要是针对大脑的功能障碍:语言发育落后,运动功能发育落后,专注力差,多动行为,睡眠障碍,的训练和治疗;也包括:情绪管理训练和心理治疗2.训练的方法通常是采用一对一训练和示范、指导的方法,这类治疗方法每天要达到一定的治疗时间量才会有显著效果。针对性的治疗的场合和地点包括:有医疗机构,专业训练机构和家庭。其中,在家庭里的由家长或治疗师上门训练是非常重要的。在医疗机构里,负责任的医生和治疗师应该在就诊和治疗的过程中指导家长如何在家庭中教育训练孩子(但是遗憾的是,由于在门诊和治疗过程时间短,效益低,资源缺乏等原因,常常无法指导家长如何在家庭正确的进行教育和训练)建议如果条件,在有经验和医生或治疗师的工作室,有足够的时间来指导和示范训练帮助家长学习教育训练的技巧,在家庭中延续在机构中做的治疗,保证有足够的针对性治疗的时间。同时,还需要指导家长预防,避免和纠正不良的、不正确的抚养和教育孩子的方法(因为这也会影响治疗的效果,更严重的会加重病情)。二、辅助对症治疗方法:治疗的项目主要包括下述方法和技术(针对不同病种和功能障碍,选用不同的治疗方法):1.理学方法治疗,主要有:脑循环,电刺激,高压氧,经颅磁治疗,生物反馈治疗2.中医治疗:针灸,埋线,推拿按摩3.药物辅助治疗(不包括促进大脑发育的神经营养添加剂。辅助治疗一般是促进大脑的功能,帮助教育训练治疗的,需要有足够的针对性治疗训练时间才能起作用。仅仅只做辅助治疗,或者大部分时间都只是做辅助治疗是不适宜的。而且,如果脑电图检查有癫痫样放电时,辅助治疗中属于创伤性,刺激性和导致兴奋性的治疗方法是要慎重或者禁止使用的。4个月龄不建议使用。在选择辅助治疗时,要因人而异,因病而异。三、支持疗法1.保证均衡营养饮食,避免挑食。2.晚上早睡,保证睡眠质量。3.使用可能促进大脑发育的神经营养添加剂。4.温馨良好的生活居住环境。是5.正确的抚养和教育方式正确的行为。6.培养良好情绪和习惯。7.音乐疗法。综上所述,在所有的治疗项目中,针对性的治疗是极为重要的,支持疗法也是很重要的,辅助治疗只是起辅助治疗目的。
广州市妇女儿童医疗中心神经内科科普号 2024年07月28日
2024年07月28日 166
166
 1
1
 2
2
-
儿童脑发育落后揭秘
北京儿童医院方铁 儿童脑发育不良,也就是儿童神经系统发育问题。这种疾病是儿童时期最严重的儿童神经系统功能障碍,困扰了大量患儿和其家属,造成患儿和其家庭陷入长期的治疗困境和误区。在这里,我尽量使用通俗的语言,为大家梳理一下这种疾病的来龙去脉,家属究竟该如何面对这种疾病。 神经系统是人体最复杂、最精密的器官和系统。在儿童时期,正是神经系统发生,发育和逐步成熟的阶段。在这个阶段,神经系统会出现各种发育相关的问题。 神经系统的发育是在特定基因表达的编码下,逐步发育的。基因的完整和功能健全,是神经系统正常发育的关键。 人类基因从父母双方各获得一半等位基因,在发育过程中,不断会有基因出现突变,也就是变异。一些变异就会影响到基因功能,出现症状,造成发育问题。因此,不是父母正常孩子就一定正常的。另外,等位基因的功能有一些是显性的表现,有一些是隐性的表现。如果出问题的基因是隐性的表现,那么只要其另一条等位基因功能正常,孩子就不出现问题,成为隐性携带者。因此,一些家长常常是疾病基因的隐性携带者,并不发病。如果父母都是问题基因的隐性携带者,那么后代中就会出现等位基因都是问题基因的情况,进而出现症状。对于家属而言,切莫认为自己家族没有疾病孩子就不会出现基因问题。 绝大多数病理性发育落后的核心原因是基因问题。目前,已经明确了几十种会造成神经系统发育问题的基因。随着科学研究的进展,还会有越来越多的问题基因被发现。对于病理性发育落后的孩子,一定要尽早进行基因检测,明确问题的具体情况,这对于诊断和后期治疗具有巨大帮助。很多家属和部分地方医院的医务人员存在认识误区:对于发育落后的孩子,认为查出来基因问题也没有帮助。这种观点是非常错误,非常落伍的。如果家属想放弃孩子,那么任何检查和治疗都不需要。如果家属不想放弃孩子,那么应该尽早明确病因,进行基因检测。 脑发育不良包括结构发育不良和功能发育不良。一些情况下,结构发育不良并不一定有功能发育不良。比如有一些孩子,胼胝体发育不好,但是功能并不受影响。反过来,很多脑功能发育不良的孩子,脑结构并不能看出问题,也就是核磁看不出问题。 一般而言,如果一个孩子,脑结构发育不良,同时临床有明确发育落后时,家属一定要带着孩子找专科医生进行病因检查,常常就是基因有问题,特别是如果脑电图也异常的情况下。 儿童发育落后可以大体分为生理性发育落后和病理性发育落后。在婴幼儿期,也就是1岁以内,很多所谓的发育落后的孩子,实际上是生理性发育落后,也就是并没有正真的问题,只是最初发育晚一些而已,就像龟兔赛跑一样,乌龟爬的慢,但是一直在前进,最后可能比兔子更早到达终点。这些孩子如果进行系统发育评估,常常是轻度发育落后,或者是发育落后一般不超过2个月以上。 生理性发育落后的孩子不需要特殊治疗。如果家属积极,可以给予孩子基本的康复训练,也就是运动康复,按摩等物理康复。绝对没有必要打针吃药和过度康复!一般而言,生理性发育落后的孩子,3个月左右的运动康复就会有明显效果。如果一个孩子,3个月左右的康复进展不大,家属一定要高度怀疑是否是病理性发育落后,尽快寻找病因。 病理性发育落后是真正意义的脑发育落后,是有明确病因的。造成脑发育落后的原因主要包括:基因问题、脑损伤以及其它疾病伴发发育落后。 基因问题是最主要的发育落后原因,不同基因问题带来的临床表现和疾病出现的时间不同,治疗方式也有差异,需要明确具体基因情况给予针对性治疗。 脑损伤,特别是严重皮层和皮层下脑软化、外伤造成軸索损伤,重度脑积水等,会造成运动障碍之外,出现认知等脑功能问题。 癫痫和一些代谢性疾病,比如线粒体病,会随着病情进展出现认知功能障碍。如果不去积极解决原发病,那么认知损伤会越来越重。 脑发育落后的治疗,不能千篇一律的单纯给予康复训练。也不要不切合实际的幻想着使用神经营养药物来解决问题。没有任何神经营养药物,能够改变脑发育,解决脑发育落后问题。 脑发育落后治疗的核心是明确病因,针对病因给予综合治疗。在明确病因的前提下,以康复训练为核心,配合针对病因的对症药物,必要时联合促进神经功能和脑网络改变的微创手术(比如迷走神经刺激和颈动脉外膜剥脱)。 脑发育从胚胎期开始,经历了漫长的时间,直至青春期停止。这个过程很漫长!同时,治疗脑发育落后也是漫长的过程,家属需要耐心和坚持。 在明确病因,给予积极的针对性的积极综合治疗下,大多数脑发育落后的孩子都会获得良好的治疗效果。让我们一起努力来改变这些孩子的命运!
方铁医生的科普号 2024年02月07日381
0
5
2024年02月07日381
0
5
-
父母要如何观察和判断宝宝语言发育落后?
语言功能是大脑发育水平和心理发展水平的一个重要指标,语言发育落后意味着孩子的智力同时也发育落后。语言是交流的工具,因此,观察宝宝的语言发育是否成熟需要在语言发育的关键年龄进行,观察和判断主要的方法如下: 2个月:用眼睛寻找声源3个月:笑出声来4个月:用头的运动来寻找声源,对人咿呀说话5个月:听到音乐发出声音或停止哭声6个月:叫时有应、发出4种不同的声音8个月:无意识地叫爸爸或妈妈、听人谈话10个月:摇头或者摇手表示"不"11个月:说两个字(清晰)且使用正确12个月:有意识叫爸爸和妈妈、清晰正确使用2个字14个月:喜欢看图片(看书30秒钟)想唱歌、叫姓名有反应15个月:对节奏声有反应,清晰正确使用4个字以音代物16个月:可识别玩具箱内的一件物品,清晰正确使用3个字18个月:翻阅图画并且对着图片说话清晰正确使用3-5个字,可识别玩具箱内的两件物品 如果10个月龄仍然缺乏眼神和动作的互动。15个月龄对简单的口头指令,叫姓名无反应。18个月还不会说3个字以上的短语,就提示语言发育落后,需要查明原因,及时干预和训练。
麦坚凝医生的科普号 2023年08月27日247
0
3
2023年08月27日247
0
3
-
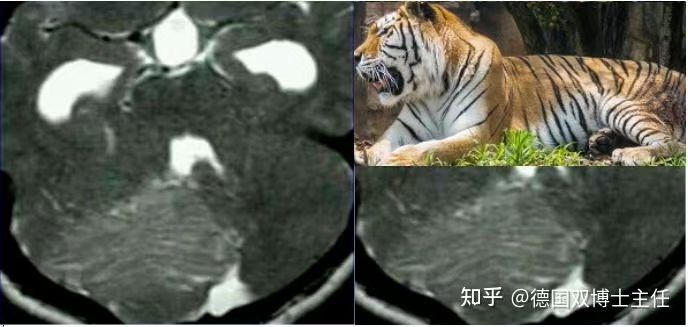
虎纹斑-小脑发育不良性神经节细胞瘤
挖呀挖,挖出“虎纹斑”:小脑发育不良性神经节细胞瘤(dysplasticcerebellargangliocytoma)又名Lhermitte—Duclos病(Lhermitte—Duclosdisease,LDD)。同行咨询病历,我的直觉是肿瘤,有占位效应。本院复查了磁共振,全国小儿肿瘤读片非常厉害[强]的李玉华老师,术前直接给了我病理诊断[强]“影像学表现深刻,看一眼就终身不忘的肿瘤”。术中发现肿瘤确实很漂亮,有赏心悦目的感觉-是不是很专(bian)业(tai)[捂脸]。良性的肿瘤,有望治愈的肿瘤。
 张晨冉医生的科普号
张晨冉医生的科普号 2023年06月26日134
0
0
2023年06月26日134
0
0
-
脑皮质发育不良,是手术好还是吃药好

 王海祥医生的科普号
王海祥医生的科普号 2023年04月11日66
0
1
2023年04月11日66
0
1
-
脑皮质发育不良,有继续发育的可能性吗?
王海祥医生的科普号2023年04月11日74
0
1
-
Fcd就是脑皮质发育不良吗?
 林久銮医生的科普号
林久銮医生的科普号 2022年11月17日121
0
0
2022年11月17日121
0
0
-
请问脑皮层发育不良必须手术吗?
 老有所依健康大讲堂
老有所依健康大讲堂 2022年09月29日241
0
1
2022年09月29日241
0
1
-
异染性脑白质营养不良(MLD)
异染性脑白质营养不良(metachromaticleukodystrophy,MLD)是一种常染色体隐性遗传性疾病,是最常见的溶酶体病,其发病是由于芳基硫酸酯酶A(aryIsulfataseA,ARSA)或神经鞘脂激活蛋白B(sphingolipidactivatorproteinB,SAP-B,saposinB)即脑硫脂激活蛋白的缺陷中,使溶酶体内脑硫脂水解受阻,而沉积在中枢神经系统(centralnervoussystem,CNS)的白质、周围神经及其他内脏组织,引起脑白质、周围神经及其他内脏组织等病变,从而产生严重代谢异常的脱髓鞘性神经系统退行性疾病。本病因为病理检查时脑白质中异常沉积的脑硫脂颗粒和蓝色的染料作用后变为红色的异染性颗粒而得名。异染性脑白质营养不良约占儿童脑白质病的8%左右。按照发病年龄,可分为经典晚期婴儿型(占50%-60%)、青少年型(占20%~30%)及成人型(占15%~20%)。遗传和流行病学MLD致病基因主要为ARSA,ARSA基因定位于22q13.33.全长3.2kb,包含8个外显子,编码蛋白含509个氨基酸。除了ARSA基因突变,也有报道PSAP基因突变导致SAP-B蛋白缺陷引起MLD,但非常少见。ARSA是一种酸性水解酶,由核糖体合成,通过甘露糖-6-磷酸依赖途径进入溶酶体内,使脑硫脂上的半乳糖3-硫酸水解脱落,而变成可溶性的小分子物质被人体再利用。ARSA基因突变可导致溶酶体内脑硫脂水解障碍,而在脑白质、周围神经及其他内脏组织内沉积,从而抑制髓鞘的形成,促进脱髓鞘的进展。体外实验表明脑硫脂沉积可引发炎症因子反应,促发细胞凋亡,导致疾病产生。异染性脑白质营养不良为常染色体隐性遗传,部分国家及地区有该病发病率数据。瑞典晚期婴儿型的发病率约为1/40000,青少年型发病率约为1/160000。在孤立的Habbanite犹太人群,该病发病率高达1/75。临床表现1.婴儿晚期型典型的婴儿晚期型疾在出生后30个月前发病,在1到7岁内逐渐死亡。第一个表现是丧失了习得的运动技能,特别是步态不稳,此时的检查显示肌张力减退,并且通常有明显的膝关节反曲,深部肌腱反射减弱甚至消失,提示神经病变。有些患者走路时间延迟,还有些也许从不会走路,但大多数患儿会独立行走,学会说简短的句子,然后这些技能就会退化。共济失调和无力可能会并发感染。最初的表现为肌张力减低和反射改变可能提示肌病或周围神经病变,腿上可能会有间歇性的剧烈疼痛。疾病进展可分为四个阶段,最初出现症状代表阶段一,在第二阶段,患者不能再站立,但可以坐,共济失调,步态不稳,构音障碍或失语,智力倒退,下肢肌张力增加,跟腱反射亢进。发展为眼球震颤,眼底镜检查显示视神经萎缩。在III期患者发生痉挛性四肢瘫。可能有去大脑或去皮质强直或有肌张力活动障碍,大约三分之一的患者发生癫痫发作,咽肌协调功能丧失,进食困难及呼吸困难,智力进一步退化,语言功能丧失。第四阶段患者不能沟通,失明,不能吞咽,此时需要鼻饲,一般死于肺炎。2.青少年型MLD一般为4到16岁之间起病,学习成绩下降,看起来不清醒或者容易走神,有些患有痴呆症,精神错乱或情绪异常。有些年纪较小的患者像婴儿型一样步态不稳。肌肉强直,姿势异常以及可能出现共济失调。发病后不到一年,患者不能行走,出现尿失禁。跟晚期婴儿型一样,进展到第三阶段和第四阶段。很明显,晚期婴儿型的病人表型可能与少年型和成年型病人表型相重叠,这种区分可能是人为的。事实上,在同一家族中,同胞兄弟姐妹可能分别被归纳为少年型和成人型。急性胆囊炎,慢性出血性胰腺炎,腹部肿块或胃肠道出血可以是这个病的特殊临床表现。3.成人型MLD成人MLD是指青春期后出现的患者。发病年龄可能早到15岁或晚到62岁。生存期可能是5年,10年甚至更长,主要表现为精神异常,可以通过神经影像学来和精神病患者区分。痴呆可能表现为记忆丧失或智力下降,精神变化可能是精神分裂症症状,情绪不稳定、焦虑或淡漠。视觉空间区分能力可能会受到损害。有患者表现为抑郁症和长期酗酒。据报道,18%和27%的患者出现幻听和妄想,53%的患者出现精神病,有患者出现抑郁症和慢性酒精中毒。运动障碍从步态笨拙发展为构音障碍,肌张力增加,跟腱反射亢进,共济失调,出现类似于帕金森的特征。后期发展为肌张力障碍,痉挛性四肢瘫,延髓受累和去皮质状态,也会出现视神经萎缩和眼球震颤,癫痫发作。疾病终末期患者失明、失语,对外界失去反应。辅助检查1.白细胞ARSA的活性 是确诊的可靠办法,该症患者不同的组织包括脑、培养的成纤维细胞、白细胞和尿液中均有酶活性的缺乏。(1)ARSA缺乏:白细胞中ARSA活性低于正常对照10%者为患者。(2)ARSA假性缺乏:是指表现正常健康而白细胞中ARSA活性水平低,为正常对照的5%~20%。在晚期婴儿型中ARSA的活性可全部缺乏,而少年型可在0%~10%之间。尿液中脑硫脂含量增多,ARSA活性降低,也可进行诊断。2.基因检测 ARSA基因和PSAP基因突变检测多用于鉴别携带者及产前诊断,并可鉴别患者基因型。3.脑脊液中蛋白质浓度的升高 脑脊液蛋白水平在婴幼儿疾病早期可能是正常的,但它浓度逐渐上升到100mg/dl或更高。这对少年早期型患者也是如此;而少年晚期型患者和成人型患者通常有正常的蛋白质水平,有少数人的浓度升高。4.影像学 计算机断层扫描(CT)或磁共振(MR)的神经成像与髓鞘水分的丢失增加是相一致的。CT低密度和磁共振成像(MRI)脑室周围的白质T2WI呈高信号、提示脑白质营养不良,后期会出现明显的脑萎缩,质子磁共振波谱提示N-乙酰天门冬氨酸的含量减少和神经胶质细胞标志物肌醇的增加。5.脑电图(EEG) 患者脑电图可能是不正常的,特别是在癫痫发作的时候,可能会有弥漫的慢波和局限的棘波发放,噪声可能引起明显的惊吓反应。成人型病人的脑电图接近是正常的。6.活检 周围神经的活检显示出有特征的包涵体。7.其他检查脑干听觉诱发反应(BAERs)、视觉诱发反应或躯体感觉反应也可能出现异常。诊断1.诊断患儿有运动智力表现倒退的临床表现,头颅MRI提示有特征性脑白质病变,芳基硫酸酯酶A活性缺乏或者检测到PSAP基因突变,即可诊断异染性脑白质营养不良。2.鉴别诊断该病需要考虑的相鉴别的是其他类型的脑白质病,如球形脑白质营养不良、多种硫酸酯酶缺乏症等,最重要的鉴别点是外周血检测到相对应的酶活性缺陷。治疗1.对症治疗如控制抽搐,使用肌松剂预防肌肉挛缩等。氨己烯酸可能有助于减少痉挛发作。2.骨髓移植青少年型及成人型在疾病早期骨髓移植有一定效果。对晚期婴儿型患者不建议骨髓移植。
付朝杰医生的科普号 2022年07月19日859
0
0
2022年07月19日859
0
0
-
克拉伯病(球形细胞脑白质营养不良、GLD)
一、什么是Krabbe病?Krabbe病(Krabbedisease),又称为球形细胞脑白质营养不良,是一由beta半乳糖脑苷脂酶(也称为半乳糖神经酰胺酶,galactosylceramidebetahydrolase,GALC)缺乏导致的溶酶体贮积病,是一种罕见的常染色体隐性遗传病。欧洲发病率估计为1/100000,美国发病率估计为1/250000,我国目前缺乏相关的流行病学资料。二、Krabbe病的致病机制是什么?Krabbe病是一种遗传病,致病基因是GALC基因,也称为半乳糖神经酰胺酶基因,位于染色体14q31,目前已识别约200种GALC突变形式。半乳糖神经酰胺酶可以使白质髓鞘形成过程中形成的半乳糖脂体发生脂质体水解。当编码半乳糖神经酰胺酶的基因发生突变时,会造成酶的活性缺陷,使半乳糖脑苷脂不能降解成神经酰酰胺和半乳糖,周围神经和中枢神经系统出现球样细胞形成和髓磷脂减少,这些病理改变和GALC缺乏时蓄积的鞘氨醇半乳糖苷和半乳糖脑苷脂的毒性有关。三、Krabbe病患者有什么临床表现?症状的严重程度和发病时间密切相关。根据发病年龄可以分为早发婴儿型(约占85%-90%),和晚发型(10%-15%)。早发婴儿期发病进展迅速,通常在2岁内去世。晚发型的症状相对轻,生存期相对更长,可以进一步分为晚发婴儿型、青少年型和成年型。早发婴儿型:症状通常可以分成三个阶段。第一阶段,在4-6个月发病之前生长良好,发病之后,出现静止不能,易激惹,呕吐,喂养困难,婴儿可能对光、声或触摸刺激异常敏感,出现强直性伸肌痉挛。第二阶段,逐渐出现视力障碍,出现对抗惊厥药物反应差的癫痫样发作。第三阶段,疾病的晚期,出现视力丧失、听力丧失和去大脑状态。晚发婴儿型:通常13-36个月龄发病,出现易激惹、视力障碍和步态异常,随着疾病进展,这些症状逐渐加重,并且出现痫性发作,情绪不稳定等。通常在6岁左右去世。青少年型:患儿逐渐出现视力障碍、震颤、步态障碍和注意力缺陷多动障碍,并且以不可预测的速度进行性加重,导致患儿严重失能,通常在诊断后的十年内去世。成年型:可表现为手灵活性丧失,无力,四肢烧灼样感觉异常,痉挛状态、共济失调、视力和听力障碍、精神发育迟滞,或可表现为肢体远端感觉丧失和肌肉萎缩,脊柱侧弯。极少患者症状局限于无力且不伴智力衰退。四、对诊断有帮助的辅助检查有哪些?影像学检查:在婴儿型Krabbe病患儿中,最常见的MRI异常累及深部脑白质、齿状核和小脑白质(图1)。在青少年型和成人型Krabbe病患者中,脑部MRI可显示出顶枕区域白质病变和/或皮质脊髓束萎缩和T2信号增高(图2),齿状核和小脑白质通常不受累。电生理检查:大多数婴儿型krabbe病患儿的运动和感觉神经传导速度均显著减慢,20%的晚发型患者中也是如此。有症状的儿童常有脑干诱发电位、视觉诱发电位和EEG异常。Krabbe病患者脑脊液脑脊液蛋白质含量通常升高,高于61.5mg/dl通常提示生存期较短。病理学检查显示周围神经脱髓鞘和过碘酸-希夫阳性多核球样细胞,中枢神经系统病理表现包括球样细胞、髓磷脂广泛性丢失,以及少突神经胶质细胞几乎完全丢失。五:如何诊断Krabbe病?通过测定劝全血中分离出的白细胞或培养的皮肤成纤维细胞中GALC活性而确诊;GALC基因检测证实Krabbe病的诊断,为遗传咨询提供依据。六、Krabbe病如何治疗?目前没有针对症状性婴儿型Krabbe病患者的对因治疗。目前有初步证据表明,如果在症状出现前进行造血干细胞移植,婴儿型Krabbe病患者可获益,但是还没有可靠的方法预测这些婴儿的较晚期表型,而且一些患者不接受治疗也仅有及轻微症状或没有症状。现有证据表明,有症状的晚发型(青少年型)Krabbe病儿童即使症状出现后进行造血干细胞移植,也是可以获益的。对症支持疗法包括:肌松剂缓解痉挛,抗惊厥药物控制痫性发作,康复治疗改善运动能力,对吞咽困难患者给予管喂营养支持等。七、Krabbe病如何预防?遗传代谢性疾病预防至关重要。预防措施包括避免近期结婚,进行遗传咨询、携带者做基因检测以及产前诊断和胚胎植入前诊断等。八、Krabbe病的预后如何?早发婴儿型的Krabbe病患者的平均寿命是13个月。大多数晚发婴儿型的患者在发病后2年内去世。青少年型和成年型的患者疾病进展速度和寿命差异较大。
 付朝杰医生的科普号2022年07月19日2499
0
0
付朝杰医生的科普号2022年07月19日2499
0
0
脑发育不良相关科普号

钟建民医生的科普号
钟建民 主任医师
江西省儿童医院
神经内科
2617粉丝299.4万阅读

马艳医生的科普号
马艳 主任医师
四川省医学科学院·四川省人民医院
儿科
7502粉丝22万阅读
麦坚凝医生的科普号
麦坚凝 主任医师
广州医科大学附属妇女儿童医疗中心
神经内科
4725粉丝30.6万阅读
-
 推荐热度5.0常杏芝 主任医师北京大学第一医院 小儿神经内科
推荐热度5.0常杏芝 主任医师北京大学第一医院 小儿神经内科小儿癫痫 20票
格林巴利综合征 3票
周围神经病损 2票
擅长:癫痫,神经肌肉病,遗传代谢病 -
 推荐热度4.3高和荣 康复师复旦大学附属儿科医院 康复科
推荐热度4.3高和荣 康复师复旦大学附属儿科医院 康复科脑发育不良 1票
新生儿疾病 1票
锥体外系疾病 1票
擅长:1. GMs、TIMP、PDMS、FMFM、GMFM等评估方法; 2. 高危新生儿神经行为筛查; 3. 新生儿超早期干预及6月龄内早期干预指导; 4. 高危儿、脑损伤、发育迟缓、脑瘫及基因代谢异常等患儿的功能评估及早期康复治疗。 -

 推荐热度4.1余建忠 副主任医师复旦大学附属儿科医院 神经外科
推荐热度4.1余建忠 副主任医师复旦大学附属儿科医院 神经外科脑肿瘤 39票
蛛网膜囊肿 12票
室管膜瘤 12票
擅长:儿童脑肿瘤(室管膜瘤、髓母细胞瘤、毛细胞星形细胞瘤,弥漫性中线胶质瘤、脉络丛乳头状癌、弥漫性高级别胶质瘤(胶质母细胞瘤)、脑干及丘脑等功能区肿瘤的显微外科手术及靶向治疗),脑积水,神经系统发育异常性疾病(蛛网膜囊肿、脊髓栓系、小脑扁桃体疝,肌张力障碍),脑血管病(烟雾病、海绵状血管瘤),病灶性癫痫(皮层发育不良FCD、 胚胎发育不良性神经上皮瘤 (DNET) 、节细胞胶质瘤、下丘脑错构瘤)等疾病的诊疗。